OVERVIEW
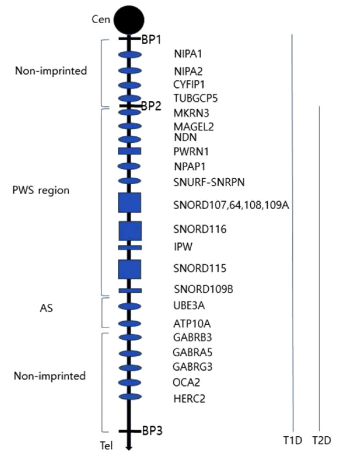
Prader-Willi syndrome (PWS; OMIM 176270) has an estimated prevalence of 1/10,000-1/30,000, and males and females are affected equally in all ethnic groups [1]. The PWS critical region is located on chromosome 15q11-q13. Normally only paternally inherited genes on this region are expressed, and PWS is caused by the abnormal expression of one or more of these genes [2,3]. Genetic mechanisms of PWS are paternal deletion of the 15q11-q13 region; maternal uniparental disomy 15; and imprinting defects [4]. Deletions in PWS are further divided into type-1 and type 2 deletion (Fig. 1).
Fig. 1.
Chromosome map of 15q11.2-13.1 region. BP1, breakpoint 1; BP2, breakpoint 2; BP3, breakpoint 3; Cen, Centromere; Tel, Telomere. Type 1 deletion (T1D) extend from BP1 to BP3, and type 2 deletion (T2D) extend from BP2 to BP3.
PWS affects multiple body systems. Prenatal growth is usually normal. Fetal movement is decreased, and there is increased need of assisted delivery or cesarean section [5,6]. At birth weight and body mass index (BMI) are 15% lower than average. Severe hypotonia during infancy can lead to poor suck and failure to thrive. Hypotonic status improve after infancy, but mild-to-moderate hypotonia persists throughout life [7,8].
As they grow up, they show hyperphagia due to lack of satiety with obsessive food seeking. They become severely obese unless food intake is strictly controlled. In uncontrolled cases, obesity, and its complications are the major causes of morbidity and mortality. Most PWS patients show developmental delay and intellectual disability [9].
Endocrine dysfunction is also common in PWS. Growth hormone (GH) deficiency is present in about 74% of cases [10,11]. GH replacement therapy has positive effects not only on growth and body composition but also on development, behavior, and nocturnal respiratory abnormalities, although a careful respiratory follow up is mandatory during long-term GH administration [12,13]. Hypogonadism is thought to have a hypothalamic origin, manifested as hypogenitalism, incomplete pubertal development and infertility [14]. Other endocrine abnormalities such as hypothyroidism, central adrenal insufficiency and type 2 diabetes can also occur [9].
MOLECULAR GENETICS AND DIAGNOSIS
The PWS region is located spans on the long arm of chromosome 15, with length of about 6Mb (Fig. 1). Within this region, at least 2.5 Mb comprises genes which are expressed differentially depending on parental origin. This region also includes several non-coding RNAs, which are believed to be involved in the regulation of alternative splicing [4,15].
Most common genetic cause of PWS patients is deletion of 15q11-q13 from the paternal origin (65-75%) [15,16]. Maternal uniparental disomy (mUPD) accounts for 20-30% of cases, which occurs when both chromosomes 15 are inherited from the mother [6,7]. The least common mechanism (1-3%) is imprinting defects (ID), caused by epimutations or micro-deletions in the PWS-imprinting center (PWS-IC).
DNA methylation analysis is usually the first step for diagnosis of PWS DNA methylation analysis can detect all three types. but it cannot distinguish the molecular class (i.e., deletion, UPD, or ID). So further investigations like FISH or CMA to determine molecular class is required. When chromosomal analysis with FISH or CMA rather than DNA methylation was first performed and reveals a deletion in the PWS/AS region (i.e., 15q11.2-q13), it is still necessary to perform DNA methylation analysis to distinguish PWS with Angelman Syndrome.
GENOTYPE-PHENOTYPE RELATIONSHIPS
There is no symptom of PWS which occurs only in specific genotype. But there are some clinical characteristics which show differences in frequency or severity according to genotype. Feeding problems, sleep disturbances, hypopigmentation and speech and language deficits are more common in deletion [17]. Type 1 deletion patients are known to have better academic performance and intellectual abilities, and more compulsiveness than individuals with type 2 deletion [18,19]. mUPD patients show higher incidence of post-term delivery, psychosis and autism spectrum disorder. On the other hand, typical PWS facial appearance or hypopigmentation are less common in mUPD patients [15].
GENES INVOLVED IN PWS
The 15q11.2-q13 region has four distinct regions divided by three common deletion breakpoints [20] (Fig. 1): 1) a proximal nonimprinted region contain four biparentally expressed genes (NIPA1, NIPA2, CYF1P1, and GCP5. 2) The “PWS region” contain five polypeptide coding genes (MKRN3, MAGEL2, NECDIN, and the bicistronic SNURF-SNRPN), C15orf2, a cluster of C/D box small nucleolar RNA genes (snoRNAs), and several antisense transcripts. 3) The “Angelman syndrome (AS) region” contain the preferentially maternally expressed genes UBE3A and ATP10A. 4) A distal nonimprinted region contain a cluster of three GABA receptor genes, the gene for oculocutaneous albinism type 2 (OCA2), HERC2 [15].
MKRN3
The Makorin Ring Finger Protein 3 (MKRN3, ZNF127) gene encodes a zinc finger protein of the Makorin family. This gene is paternally expressed. This gene is known to inhibit initiation of puberty, and loss of function mutations in MKRN3 are recognized as the main genetic cause of central precocious puberty [21]. MKRN3 is thought to be related with hypogonadism and infertility in PWS.
MAGEL2
Hyperphagia is commonly observed in individuals with PWS, associated with a defect in the hypothalamic arcuate nucleus. Interaction of neuropeptide Y (NPY) and agouti-related peptide (AgRP) stimulates food intake, whereas proopiomelanocortin (POMC) reduces it. Loss of MAGEL2 expression inhibits POMC neurons, thus food intake is less repressed [22,23]. But patients with point mutations on the paternal allele of MAGEL2 does not usually show the high appetite and severe obesity though other clinical characteristics of PWS are present, which can disassociate MAGEL2 and the hyperphagia.
Loss of expression of MAGEL2 also impairs reproductive function in mice. MAGEL2-null females showed extended and irregular estrous cycles, and males displayed decreased testosterone levels, and reduced pheromone detection. These results suggest that lack of expression of MAGEL2 contributes to the reproductive deficiencies observed in PWS.
NDN
The Necdin (NDN) gene encodes a DNA binding protein necdin, which is postulated as a key regulator of GnRH levels both in vitro and in vivo, modulating essential intracellular processes for neurite and axonal outgrowth [24-26]. Also, NDN paternal-deficient mice showed irregular breathing and sleep apneas. NDN might be a genetic factor contributing to apneas and respiratory dysfunctions of PWS.
lncRNAs
Within the Prader-Willi region there are a series of long non-coding RNAs (lnc RNAs) which are characteristically more than 200 nucleotides long. They are thought to be involved in epigenetic modifications of DNA, and regulation of gene expression at transcriptional and post-transcriptional levels [27-29]. One of the lnc RNAs, Prader-Willi Region Non-Protein Coding RNA 1 (PWRN1), is biallelically expressed in the testis and kidneys, and monoallelic expressed in the brain [30]. PWRN1 is thought to have an indirect role on the imprinting mechanism through keeping the paternal allele in an open chromatin configuration, allowing assess to transcription factors [31].
NPAP1
The Nuclear Pore Associated Protein 1 (NPAP1) is biallelically expressed in adult testis and monoallelically expressed in fetal brain, including the hypothalamus which is related to several endocrine features of PWS [30,32]. This gene is also associated with the Nuclear Pore Complex (NPC), in which the main function is to regulate macromolecular transport between the nucleus and the cytoplasm. NPCs also participates in several nuclear processes, such as gene regulation, mRNA biogenesis, and cell cycle control.
SNURF-SNRPN
SNURF-SNRPN is a complex bicistronic gene encoding two different proteins. SNURF is encoded by exons 1-3 and produces a small nuclear protein of unknown function [33], exons 4-10 correspond to the SNRPN portion and encode the protein SmN, involved in mRNA splicing [34].
Small Nucleolar RNAs (snoRNAs) are within the SNURF-SNRPN transcript. snoRNAs participate in DNA methylation, alternative splicing and post-transcriptional regulation [4,35]. The PWS region encompasses five single copy snoRNA genes (SNORD64, SNORD107, SNORD108, SNORD109A, and SNORD109B) and two snoRNA gene clusters (SNORD115 and SNO-RD116). Although most of the SNORDs are ubiquitously expressed in human tissues, SNORD115 and SNORD109B are expressed exclusively in the brain.
SNORD116 has emerged as a critical, and possibly, determinant candidate in PWS. Patients with small deletions (150-200 kb) or translocations showed PWS phenotype [36-38]. Snord116-KO mice also displayed PWS features such as post-natal growth retardation and hyperphagia [39,40]. Mice model SNORD116 was knocked-out specifically in NPY neurins in the hypothalamic arcuate nucleus showed the same overall phenotype observed in mice lacking SNORD116 globally; low birth weight, increased body weight gain in early adulthood, increased energy expenditure and hyperphagia [41]. This suggests that SNORD116 control NPY neuronal functions, and thus food intake and energy homeostasis. Also, Snord116-deficient mice showed decreased activity of the hypothalamic prohormone convertase PC1 impairing the prohormone processing of proinsulin, pro-GH-releasing hormone, and proghrelin [42]. In addition, Snord116-deficient mice displayed loss or shift in methylation dynamics of CpG islands in the cerebral cortex dependent on the circadian cycle [43].
SNORD115 gene encodes the serotonin receptor 5-HT2C [35]. 5-HT2C gene encodes G protein-coupled receptor specific to the brain, whose activation is associated with a variety of physiological processes, such as dopamine modulation, anxiety, sleep regulation, satiety response, energy balance, and locomotor activity [44]. Lack of SNORD115 is not sufficient to cause PWS, but a phenotypic effect when absent along with other genes in the PWS critical region cannot be excluded. 5-HT2C receptor knockout mice that developed hyperphagia and late-onset obesity [45,46]. The absence of SNORD115 expression in PWS accompanied by the impairment of the 5-HT2C receptor activity may be partly responsible for some of the behavioral and metabolic features of PWS.
EPIGENETICS
Maternal microdeletions of the Angelman syndrome imprinting control region (AS-ICR) I causes Angelman syndrome [47, 48]. The AS-ICR contains alternate 5´ noncoding exon for SNRPN that are uniquely expressed in oocytes [48,49]. Oocyte-specific transcription leads to methylation and transcriptional silencing of the maternal allele of the PWS-ICR. In addition, some individuals with AS have imprinting mutations that deletes a binding site for the transcription factor SOX2 [50]. These studies of AS imply that AS-ICR has an important role for establishing silencing the maternal allele of the imprinted genes within the PWS locus.
There are several additional epigenetic marks differentiated by parental origin at the PWS-ICR. Histone H3 lysine 9 (H3K9) methyltransferase SETDB1 associates with the transcription factor ZNF274 bound to sites within the 5´ cluster of SNORD116 repeats [51]. Knockdown or inhibition of either SETDB1 or ZNF274 induced SNORD116 transcript expression from the normally silent maternal allele [51-53].
Epigenetic Therapies
The general strategy for epigenetic strategies for PWS involves de-repressing the maternal silent PWS-ICR to activate SNRPN and SNORD116 transcription [54]. Several inhibitors of EHMT2/G9a, a histone 3 lysine 9 methyltransferase were able to reactivate the expression of paternally expressed SNRPN and SNORD116 from the maternal chromosome, both in cultured PWS cell lines and in a PWS mouse model [55,56]. Inhibition of SETDB1 using shRNA knockdown resulted in partial reactivation of SNORD116 and 116HG in PWS-derived iPSC cell lines and neurons [51]. In addition, when inactivated ZNF274 using CRISPR/Cas9 in PWS-derived iPSC lines, reactivation of both SNRPN and SNORD116 resulted, as well as a reduction of H3-K9me3 at the PWS-ICR [53]. These studies suggest that epigenetic strategies for unsilencing maternal SNORD116 are potential candidates for treatments of PWS.
GENETIC COUNSELING
Recurrence risk of PWS differs according to genotype. Sporadic deletion cases have < 1% risk of recurrence, while recurrent risk in structural abnormalities of chromosome 15 (such as translocations, ring formation, isochromosome or inversions) can be as high as 25-50%.
Occurrence of mUPD 15 is typically de novo and recurrence risk is low (< 1%). However, there is possibility that the father has a Robertsonian translocation involving chromosome 15. This can result in a sperm nullisomic for 15, which would result in an embryo with mUPD 15. So, the father of the child with mUPD should be recommend to perform chromosomal analysis.
Rarely, a small marker chromosome is present in a patient with mUPD 15 [57]. In these patients, both parents’ karyotype should be examined, as these small marker chromosomes may increase the risk for nondisjunction and UPD [58].
The majority (approximately 85%) of PWS patients with an ID have a de novo epigenetic mutation (IIIb) with the recurrence risk below 1%. However, in approximately half of these with microdeletion in the IC (IIIa), the IC deletion is familial and the recurrence risk is 50%. Therefore, for patients with imprinting defects, IC analysis by MS-MLPA (Methylation-Specific Multiplex Ligation-dependent Probe Amplification). MSMLPA or DNA sequencing should be performed to investigate the exact origin [1,59]. And fathers of patients with an IC deletion should be recommended with DNA methylation and dosing analysis (or sequence analysis) [15].