Introduction
Various molecular techniques have significantly advanced the conservation, restoration, and habitat survey research of wildlife, including endangered and rare species. Genetic analysis, involving indoor propagation and lineage management, is integral to ex-situ conservation strategies for protecting endangered species (Kelly, 2010). Moreover, a molecular species identification method has been developed to differentiate related and endangered species that are morphologically indistinct and to explore their habitats (Cha et al., 2023).
In insects, genomic DNA (gDNA) extraction for genetic analysis, including population genetics and species identification, typically requires non-lethal sampling. This involves collecting small tissue samples, which may mildly damage the target species (Rose et al., 1994; Lushai et al., 2000; Starks & Peters, 2002; Hamm et al., 2010). However, in many insects, particularly those with small body sizes, this method of tissue collection for gDNA extraction can negatively impact their physiology and behavior (Scriven et al., 2013). Consequently, this approach to gDNA extraction might contribute to the population decline in endangered insects with already small populations.
Polyphylla laticollis manchurica (PLM) belongs to the coleopteran family Scarabaeidae and is classified as Critically Endangered (CR) in the Red Data book of the Republic of Korea, as indicated by the National Institute of Biological Resources (2023). Additionally, PLM has been designated as a class I endangered species by the Ministry of Environment of South Korea since 2005. Previously, PLM was widespread along rivers throughout South Korea. Currently, their distribution is confined to limited areas, specifically along the Geumgang River (Cheongju-si, Buyeo-gun, Nonsan-si, and Sejong-si). The developmental period of the beetle, from larva to adult, spans 3-4 years. Therefore, larvae living under reed beds are vulnerable to flooding during the rainy season and to urban river development. Since the larval stage involves residing underground, verifying the presence or absence of PLM within their habitat outside the adult stage is challenging. Moreover, differentiating PLM larvae from those of related species coexisting in the habitat using morphological species identification methods is difficult without expertise. Incidentally, this challenge can be addressed using molecular species identification methods. However, since the larvae’s body consists of easily damaged skin and legs that are crucial for digging and moving underground, removing these parts for gDNA extraction would adversely affect larval survival.
Over the past two decades, several noninvasive gDNA sampling methods have been developed, enabling species identification and genetic analysis without causing physical damage to the target species (Waits & Paetkau, 2005). These methods have been predominantly applied to species that are challenging to capture due to their exceedingly small populations or restrictive conservation policies. In these instances, researchers have extracted gDNA for species identification and genetic studies from sources such as egg shells (Storer et al., 2019), feces (Fumanal et al., 2005; Lefort et al., 2012; Scriven et al., 2013; Sint et al., 2015), mucus (Armbruster et al., 2005; Palmer et al., 2008), and molted skin (Inoda et al., 2015; Kranzfelder et al., 2016; Ali et al., 2019; Sittenthaler et al., 2023; Kim et al., 2024).
Recently, researchers have conducted noninvasive species identification studies on Coleoptera using molted skin (exuviae) from diving beetle larvae (Dytiscidae) (Inoda et al., 2015) and feces from adult weevils (Curculionidae) (Fumanal et al., 2005) and scarab larvae (Scarabaeidae) (Lefort et al., 2012). However, in research on the feces of scarab larvae, samples collected after feeding carrots without soil under laboratory conditions were used for gDNA extraction, which does not reflect their natural underground environment that typically contains soil and digestive matter.
However, despite the development of several non-invasive species identification methods using feces, the PCR success rate has been relatively low compared to when other non-tissue materials such as exuviae and mucus are used. This reduced efficiency is attributed to the low initial amount of gDNA in the feces or its degradation by nuclease activity (Sint et al., 2015), and the presence of PCR inhibitors that are co-extracted with the gDNA from the feces (Fumanal et al., 2005; Ali et al., 2019).
This study initially aimed to determine whether feces from PLM larvae could yield gDNA samples suitable for noninvasive species identification using species-specific markers. The second objective was to enhance the previous direct gDNA extraction method (Cha et al., 2020) by addressing the challenges of low DNA quantity and PCR amplification inhibition using a rapid, environmentally friendly, and cost-effective gDNA extraction method.
Materials and Methods
Beetle larvae collection and rearing
PLM is classified as a class I endangered species by the Ministry of Environment of South Korea. Consequently, capture and storage were undertaken only after securing permission from the Geum River Basin Environmental Office (Permission No. 2023-25).
To collect PLM larval feces, male and female adult beetles were captured in Cheongju, South Korea (36°39′04.0″N, 127°22′31.0″E) on July 14, 2023, by using a light trap. A pair of male and female adults were placed in a rearing case (acryl case with a breathable lid: 30 cm high, 20 cm wide) filled with river sand to a 20 cm height for mating. Oak leaves were cut and provided as food until eggs were laid; the eggs were collected and stored individually in Petri dishes (10 cm diameter) filled with river sand where they hatched.
The larvae were reared in a case containing wheat seedlings in a soil substrate mixed with fermented sawdust and river sand at a 2:1 ratio. Wheat roots and soil substrate were provided as food for the larvae and replaced every 2 weeks before the roots dried out. All rearing cases for larvae and adults and Petri dishes for hatching were kept at 24 ± 2°C and 50-60% relative humidity (RH), under a 16:8 h light and dark photoperiod.
Beetle larvae for feces collection were gathered from the sympatric habitat of PLM at the following locations: Cheongju-si (36°39′33.3″N, 127°22′54.8″E) and Sejong-si (36°34′51.7″N, 127°18′20.0″E). All coexisting beetle larvae were extracted by digging under the reed bed in the PLM habitat and were reared under the previously described conditions.
DNA extraction from larval feces
All PLM larval feces were collected from 2nd instar larvae after their transfer from burrows in the rearing case to a 1.5 mL tube with featherweight forceps 1 day after exchanging wheat seedlings and soil substrate. To avoid cross-contamination, the forceps were sterilized with 100% ethanol after each collection. The weight of a single piece of 2nd instar PLM larval feces was estimated at approximately 13-15 mg. In the same manner, feces from coexisting beetle larvae (unknown instar stages) were also collected; all feces were then stored at −20°C until gDNA extraction.
In this study, both the direct gDNA extraction method (DAP buffer; 20 mM sodium hydroxide, 5% polyethylene glycol 200, and 5% dimethyl sulfoxide) (Cha et al., 2020) and the silica-membrane gDNA extraction method (DNeasy Blood & Tissue kit; Qiagen, Hilden, Germany) were employed to compare the fecal gDNA extraction efficiency. Initially, gDNA extraction using the DAP buffer was optimized as follows: two pieces of feces (approximately 30 mg) were placed into a 1.5 mL tube, followed by the addition of 300 μL of DAP buffer. The feces were then thoroughly homogenized using a plastic pestle, after which the tube was incubated at 23-25°C for 10 min, with the sample being mixed three times to ensure thorough mixing of debris and DAP buffer. After complete separation of fecal debris and supernatant, 5 μL of supernatant was transferred to a new 1.5 mL tube, to which 995 μL of nuclease-free water was added (200-fold dilution factor). This diluted sample (5 μL) was then used as the PCR template. Both the stock supernatant and diluted solution were stored at −20°C for subsequent experiments.
Secondly, the gDNA extraction using the DNeasy Blood & Tissue kit was performed according to the manufacturer’s instructions with slight modifications. Specifically, two fecal pieces were transferred into a 1.5 mL tube containing 270 μL ATL buffer and 30 μL proteinase K solution. The feces were completely ground using a plastic pestle, followed by incubation at 56°C for 1 h. The subsequent steps were conducted as per the manufacturer’s guidelines, concluding with a final elution of 25 μL. The eluted solution (5 μL) served as the PCR template, and the remaining solution was stored at −20°C for future use.
Primer design and PCR procedure
Given that gDNA derived from non-tissue sources (feces, exuviae, mucus, etc.) is often fragmented, we aimed to identify an optimal species-specific marker. To achieve this, we developed species-specific markers based on amplicon size, resulting in four pairs of markers designed and validated using the cytochrome b gene (Genbank accession number: KF544959.1) of PLM (Fig. 1). Each primer was crafted by comparing it to cytochrome b genes from three related scarab beetles (Popillia mutans, NC056126.1; Mimela splendens, MZ064554.1; Holotrichia niponensis, MZ726798.1) through alignment. We then compared the amplification efficiency of gDNA from PLM larval feces to select the final species-specific marker (Fig. 2). Unlike findings by Fumanal et al., (2005), our study demonstrated that larger amplicons were more effectively amplified than smaller targets of the cytochrome b gene when DAP buffer was used for gDNA extraction from PLM larval feces.
The primer pair PLM_CytB_SF_F1 and PLM_CytB_SF_R1, which generated an 815-bp fragment of the cytochrome b gene from PLM, was chosen. The selection was based on the fact that the longer base sequences provide more data to verify sequence variation between individuals, which is valuable in population genetic studies.
PCR amplification employed the primer sequences PLM_CytB_SF_F1 (forward) 5′-TTAATTGACCTCCCCTCAC-3′ and PLM_CytB_SF_R1 (reverse) 5′-GTTTATTGGGGATAGAGCGC-3′, along with AccuPower® PCR premix (Bioneer, Daejeon, South Korea) following the manufacturer’s guidelines. In summary, the PCR was conducted in a 50 μL reaction volume containing 5 μL gDNA solution extracted by DAP buffer or DNeasy Blood & Tissue kit, 10 pmol each of forward and reverse primers, 42.6 μL nuclease-free water, and an enzyme pellet tube. The components in the pellet tube were thoroughly mixed until the enzyme was entirely dissolved. The thermal cycling protocol was as follows: initial denaturation at 95°C for 1 min, followed by 50 cycles at 95°C for 30 s, 58.5°C for 30 s, and 72°C for 1 min, concluding with a final extension at 72°C for 3 min. The PCR products underwent gel electrophoresis to verify successful amplification. To assess nucleotide variations within the gene fragment, residual PCR products were purified using a QIAquick PCR purification kit (Qiagen) and sequenced at Macrogen (Seoul, South Korea) to detect sequence variation among individual samples. All partial sequences of the cytochrome b gene from PLM larval feces were registered in GenBank under accession numbers PP112617-PP112626.
Optimization of direct DNA extraction method
Feces contain various substrates, including humic acids from the soil, nucleases from target species, microorganisms native to the soil, and digestive residues. Particularly, humic acid in soil is known to inhibit PCR amplification (Braid et al., 2003; Schrader et al., 2012). Therefore, we aimed to optimize the direct extraction method by adjusting the feces amount and the dilution factor to minimise substances that could inhibit the PCR reaction. To establish the optimal ratio of DAP buffer to feces for gDNA extraction, gDNA was extracted using varying buffer amounts based on feces weight (30 mg of feces per 300 μL; 60 mg of feces per 600 μL; 90 mg of feces per 900 μL; 120 mg of feces per 1,200 μL), and the PCR amplification efficiency of each was then compared (Fig. 3A). Next, to identify the optimal dilution factor, we diluted the feces and DAP buffer mixture lysate at various ratios (from 50- to 400-fold), and subsequently compared the PCR amplification efficiency (Fig. 3B). In optimizing the dilution factor, 30 mg and 300 μL of feces and DAP buffer were used, respectively. All PCR amplification procedures for optimizing the direct gDNA extraction method were conducted as previously described.
Specificity verification of the species-specific marker
To confirm the specificity of the species-specific marker, PCR amplification using gDNA from the feces of PLM larvae and other sympatric scarab beetle larvae (CJ from Cheongju-si; SJ-1 and SJ-2 from Sejong-si) was performed. This verified whether a non-target amplicon was produced due to coexisting beetles. Direct gDNA extraction was carried out using the optimized gDNA extraction conditions (300 μL of DAP buffer with the feces weight adjusted to 30 mg), presuming the experiment maintained consistent gDNA concentration. The remaining procedures were consistent with those previously described.
Furthermore, to identify the sympatric species involved in this experiment and compare the results with the specificity verification of the species-specific marker, species identification was conducted using universal primers. For this, gDNA was extracted from larval legs, and a pair of universal primers (LCO1490, HCO2198) was employed to amplify the cytochrome c oxidase I (COI) fragment (Folmer et al., 1994). The PCR procedure consisted of a denaturation step at 95°C for 3 min, followed by 40 thermal cycles of 95°C for 30 s, 57°C for 30 s, and 72°C for 1 min, ending with a final elongation step at 72°C for 3 min. The PCR amplicons of the COI fragments from the three sympatric coleopteran species were sequenced and submitted to GenBank under accession numbers PP126446-PP126448.
Measurement of limit of detection time of gDNA in larval feces
The feces were exposed to an artificial environment that mimicked PLM’s natural habitat to determine the duration of fecal gDNA detection by the PCR amplification process. All feces were uniformly collected within one day of excretion from individual larvae. Each sample, consisting of 30 mg of feces (two pieces from 2nd instar larvae), was placed in a cell culture dish (4 cm diameter × 1 cm height) filled with a soil substrate composed of fermented sawdust and river sand in a 2:1 ratio. The dishes were then incubated at 24 ± 2°C with 50-60% RH under darkroom conditions for 30 days. Subsequently, fecal gDNA extraction was carried out at 5-day intervals for PCR amplification, with the process repeated thrice at each interval.
Results and Discussion
Selection of optimal species-specific marker pair and gDNA extraction method
Genomic DNA is well preserved under certain conditions in nature and is the fundamental material for genetic research, including the study of ancient and environmental DNA (Millar et al., 2008; Thomsen & Sigsgaard, 2018). However, gDNA can degrade rapidly due to factors like ultraviolet radiation, high temperatures, and enzymes such as nuclease. Identifying a gene sequence region that remains well-preserved in nature is thus crucial for designing species-specific markers using materials like feces and exuviae exposed to environmental conditions for specific periods. In our study, we observed that fecal gDNA could be degraded by various nucleases or intestinal microorganisms present in feces before and after excretion. Hence, we selected the well-conserved PLM cytochrome b sequence region as likely suitable for PCR amplification using the direct gDNA extraction method (Fig. 1).
Contrary to assumptions, PCR amplification of fecal gDNA using four primer pairs (Table 1) revealed that amplification reaction efficiency for small-sized PCR products was not higher than that for large-sized PCR products under identical PCR conditions. This efficiency tended to increase with the size of the PCR product (Fig. 2). All primers for cytochrome b from PLM were designed to have the same annealing temperature (57.6 ± 0.6°C), so differences in amplification efficiency were unlikely due to annealing temperature variations. Although prior research using noninvasive species identification from feces suggested that large-sized PCR products could have lower amplification efficiency due to gDNA degradation (Fumanal et al., 2005), our results indicated that this assumption might not hold under certain conditions.
In addition to comparing the PCR amplification efficiency of each primer pair, we anticipated that obtaining more sequences would enhance the available data on genetic diversity among PLM individuals. Consequently, we selected a pair of primers, PLM_CytB_SF_F1 (forward) and PLM_CytB_SF_R1 (reverse) (Fig. 1), which amplify the 815-bp fragment of the cytochrome b gene as a species-specific marker for PLM feces.
Moreover, the gDNA extraction results from the two methods exhibited significant differences. Using the direct gDNA extraction method (DAP buffer), the lysate solution extracted from feces yielded PCR amplification products that indicated successful amplification of all target gene regions. Conversely, using the silica-membrane gDNA extraction method (DNeasy Blood & Tissue kit), the extraction of the eluted solution from feces generated no PCR amplification products under any conditions with four primer pairs (Fig. 2). This lack of amplification is attributed to the composition of feces from scarab larvae, which include soil and digestive debris containing humic acid and polysaccharides that inhibit PCR and co-extract with gDNA (Monteiro et al., 1997; Schrader et al., 2012).
Although the direct extraction method with DAP buffer involves a dilution process, it is likely that some humic acids and polysaccharides remain in the lysate. Nonetheless, the occurrence of the PCR amplification reaction using the lysate suggests that the presence of these substances in the eluted solution from the silica-membrane method does not directly inhibit amplification. It is hypothesized that impurities derived from feces alter the buffer’s pH, disrupting the electronic bonds formation between silica and DNA and preventing DNA from binding to the membrane (Vandeventer et al., 2012). Despite these hypotheses, further studies are necessary to conclusively determine the causes.
Optimization of direct gDNA extraction method
To optimize the DAP buffer-based direct gDNA extraction method for fecal gDNA, we selected the best combination of fecal weight and dilution factor, considering the corresponding PCR amplification efficiency. gDNA content in feces is extremely low (Sint et al., 2015), and can include symbiotic or environmental microorganisms post-excretion. Thus, measuring total gDNA does not reflect the true amount of target species DNA, and PCR amplification efficiency could be influenced by the presence of fecal PCR inhibitors like humic acid and polysaccharides.
For the above reasons, we first aimed to determine the optimal quantity of feces for direct gDNA extraction, assessing PCR amplification efficiency as influenced by fecal quantity. We hypothesized that PCR amplification efficiency would increase with the extraction of more gDNA from larger quantities of feces; however, amplification was suppressed beyond a certain fecal threshold (Fig. 3A). This suppression is likely due to increased PCR inhibition associated with higher concentrations of PCR inhibitors in the lysate solution. The dilution step in the direct gDNA extraction method is designed to minimize impurities in the lysate solution that may impede PCR amplification. However, a higher dilution factor corresponds to a lower final gDNA concentration. Accordingly, to optimize the dilution factor to minimize PCR inhibitors while maintaining adequate gDNA concentration for PCR amplification, the lysate solution was diluted between 50- and 200-fold, and the resulting PCR amplification efficacy was compared (Fig. 3B).
A 50-fold dilution factor completely suppressed the PCR reaction, yielding no PCR amplicons. Meanwhile, a 400-fold dilution factor slightly decreased the quantity of PCR amplicons relative to those obtained with the 100-200-fold dilution factor. These results suggest that at low dilution ratios, high concentrations of impurities continue to inhibit the PCR reaction, whereas at high dilution ratios, the final gDNA concentration is reduced. Additionally, since the lysate solution used in the DAP buffer-based direct gDNA extraction method could proceed to the amplification reaction without dilution (Cha et al., 2020), the inhibition observed at a 50-fold dilution is unlikely to be due to the high pH of the DAP buffer.
Considering the results discussed, the optimized DAP buffer-based direct gDNA extraction method demonstrated optimal PCR amplification efficiency when 10 mg of feces was used per 100 μL of DAP buffer, diluted to 200-fold. Previous studies on noninvasive gDNA sampling, including the direct gDNA extraction method, document gDNA extraction from insect feces. These studies, however, faced challenges such as the failure to amplify long target gene fragments (Fumanal et al., 2005), significantly reduced amplification efficiency (Scriven et al., 2013), or amplification inhibition (Ali et al., 2019), likely due to the same PCR inhibitors co-extracted with fecal gDNA. The direct extraction method proposed here may offer an alternative solution.
Verification of the specificity of the species-specific marker
To the naked eye, scarab larvae display remarkably similar external morphology in color and shape. Therefore, distinguishing between scarab larvae without taxonomy key information is challenging (Bain, 1980). However, until recently, the taxonomy key for the larvae of several scarab beetles was difficult to employ without dissection. To address the difficulties associated with noninvasively distinguishing between target and sympatric species, the present study employed fecal gDNA and species-specific markers without the need to obtain larval tissue (Fig. 4A).
The 815-bp PLM cytochrome b fragment exhibited high levels of target specificity as a species-specific marker and only amplified PLM gDNA (Fig. 4B). This indicates that the fecal gDNA species-specific marker can discriminate between target and sympatric species coexisting within the habitat during the larval stage, when morphological distinction is challenging. Additionally, to obtain accurate species information on sympatric scarab larvae for which the morphological classification key is unknown, the COI sequence was analyzed and compared with BOLD Identification System data (Ratnasingham & Hebert, 2007). Consequently, the three sympatric larvae, CJ, SJ-1, and SJ-2, in this study were classified as CJ; Bifurcanomala aulax (Wiedemann) (GenBank accession number: PP126446), SJ-1; Blitopertha pallidipennis Reitter (GenBank accession number: PP126447), and SJ-2; B. pallidipennis Reitter (GenBank accession number: PP126448). As a result, the noninvasive species identification method using PLM feces and species-specific markers facilitates rapid species identification by eliminating the need for time-consuming and costly sequencing.
Therefore, the method proposed here could confirm the presence or absence of target species in their habitat as well as during the adult period. Furthermore, rapid noninvasive species identification could enable prompt policy decisions to preserve the habitats of endangered species in urban areas without impacting their populations.
Determination of the detection limit for gDNA in feces
To measure the residence time of fecal gDNA, PCR amplification was performed after exposing feces to artificial environmental conditions. PCR amplification at 5-day intervals up to 30 days confirmed that fecal gDNA remained detectable long enough to enable a reaction (Table 2). These results suggest that fecal gDNA can persist longer than previously reported (Lefort et al., 2012), and scarab larvae feces may serve as valuable gDNA samples for noninvasive species identification in habitats. However, since the detection probability on the 15th day was lower than on the 20th day, it appears that the remaining gDNA in the feces does not decrease proportionally over time. Therefore, when using feces for species identification, it is advisable to perform PCR at least three times per feces sample to ensure result reliability.
Conclusions
In summary, this study developed and verified a noninvasive species identification method for PLM larvae using species-specific cytochrome b markers and a direct extraction method. Compared to the previous method of obtaining scarab larvae fecal gDNA, the proposed method offers the advantage of rapid and cost-effective fecal gDNA extraction using DAP buffer, eliminating the need for commercial gDNA extraction kits. The application of species-specific markers also reduces the time and expense involved in determining the presence or absence of target species in their habitats by eliminating the sequencing step. Furthermore, given the prolonged preservation of PLM larvae fecal gDNA, fecal collection emerges as a crucial tool for confirming the presence or absence of target species within habitats. Collectively, this method’s quick species identification capability could serve as a preliminary monitoring tool for preserving PLM habitats in urban areas at risk from development. Additionally, non-invasive species identification methods that utilize residual biological materials, such as feces, secretions, and mucus, provide a means to conduct genetic studies on endangered insect species with minimal physical impact.
Acknowledgments
This research was supported by the study on population reinforcement and restoration of endangered insects and invertebrates (NIE-B-2024-48) from the National Institute of Ecology (NIE), Republic of Korea. We would like to express our gratitude to Mr. Jongdae Park for his assistance in capturing adult beetles and collecting PLM eggs, and to Mr. Mannyun Kim for his help in collecting coexisting beetles within their habitats.
Author Contributions
All authors contributed equally to the conceptualization, data curation, analysis, and experimental methodology of this study, and collaboratively wrote, edited, and reviewed the draft.
Figures and Tables
Fig. 1.
Nucleotide sequence alignment of cytochrome b from Polyphylla laticollis manchurica and other closely related scarab beetle species. The position of the five primers for the selection of optimal species-specific markers is illustrated using varied colored boxes: PLM_CytB_SF_F1, red; PLM_CytB_SF_R1, green; PLM_CytB_SF_R2, light cyan; PLM_CytB_SF_R3, orange; PLM_CytB_SF_R4, magenta. The cytochrome b sequences of one target species and three scarab beetles were sourced from GenBank, with the names and accession numbers as follows: Polyphylla laticollis manchurica (PLM, KF544959.1), Popillia mutans (PM, NC056126.1), Mimela splendens (MS, MZ064554.1), and Holotrichia niponensis (HN, MZ726798.1). Sequence dissimilarity is highlighted by blue-colored gradations.
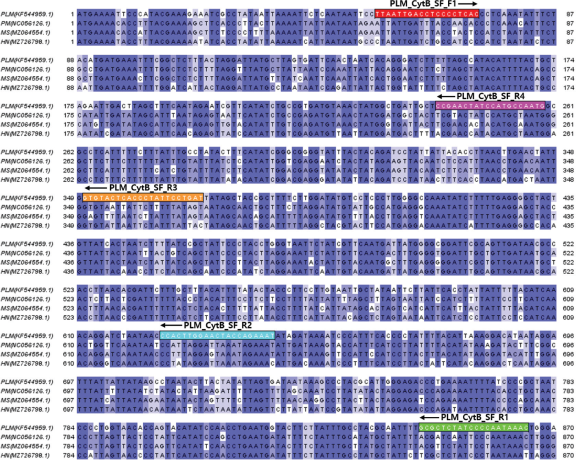
Fig. 2.
Comparison of PCR amplification efficiency of species-specific marker pairs and fecal gDNA for extraction method selection. Primer pairs producing amplification products less than 500 bp exhibited lower amplification efficiency than those yielding products over 500 bp under the same conditions. DAP and B&T for selected fecal gDNA extraction methods indicate that the direct gDNA extraction method utilizes DAP buffer, and the DNeasy Blood & Tissue kit of QIAgen, respectively. The indicated 1st to 4th pairs refer to the following primer pairs for optimal species-specific markers selection: 1st pair (PLM_CytB_SF_F1 and PLM_CytB_SF_R1), 2nd pair (PLM_CytB_SF_F1 and PLM_CytB_SF_R2), 3rd pair (PLM_CytB_SF_F1 and PLM_CytB_SF_R3), 4th pair (PLM_CytB_SF_F1 and PLM_CytB_SF_R4). Mk: 100 bp DNA ladder size marker.
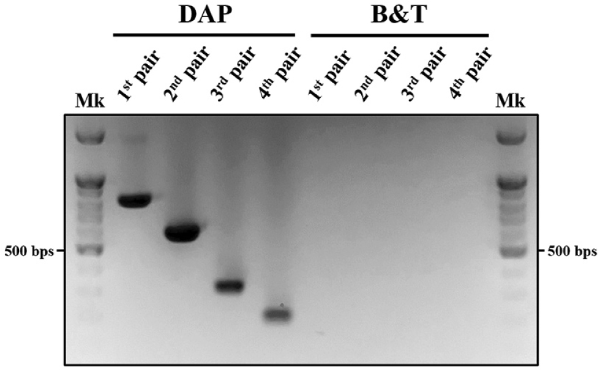
Fig. 3.
Comparison of PCR amplification efficiency according to feces amount and dilution factor for establishing the optimal direct gDNA extraction method. The optimal combination of feces amount and dilution factor for the direct gDNA extraction method was determined to be 60 mg and a 200-fold dilution, respectively. The figures demonstrate the PCR amplification efficiency relative to the amount of feces used (A) and the dilution factor (B). Black arrowheads indicate the target PCR amplicons of cytochrome b. Mk: 100 bp DNA ladder size marker.
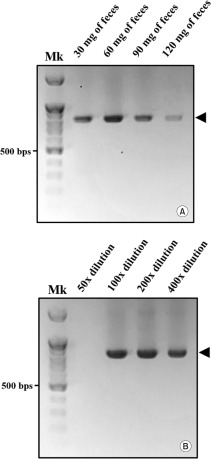
Fig. 4.
The Specificity of the species-specific marker for Polyphylla laticollis manchurica cytochrome b. To verify the specificity of the species-specific marker, we compared the results of PCR amplification using target species and sympatric scarab beetle larvae feces. (A) External morphology of the larvae and feces used in the experiment. PLM, the larva of Polyphylla laticollis manchurica; CJ, the larva of sympatric beetles from Cheongju-si; SJ-1 and SJ-2, the larvae of sympatric beetles from Sejong-si. (B) PCR amplification reaction results of gDNA extracted from the feces of each larva. Black arrowheads indicate the target PCR amplicons of cytochrome b. Mk: 100 bp DNA ladder size marker.
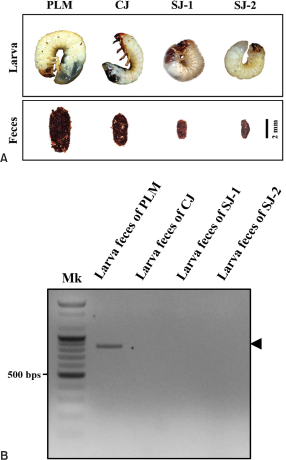
Table 1
PCR amplification of fecal gDNA using pairs of primers
| Pair no. | Pairs of primers | Sequence | Product size | |||
|---|---|---|---|---|---|---|
| 1st pair | PLM_CytB_SF_F1 | 5′-TTAATTGACCTCCCCTCAC-3′ | 815 | |||
| PLM_CytB_SF_R1 | 5′-GTTTATTGGGGATAGAGCGC-3′ | |||||
| 2nd pair | PLM_CytB_SF_F1 | 5′-TTAATTGACCTCCCCTCAC-3′ | 654 | |||
| PLM_CytB_SF_R2 | 5′-ATTTCTGGTAGTTCCAAGTGG-3′ | |||||
| 3rd pair | PLM_CytB_SF_F1 | 5′-TTAATTGACCTCCCCTCAC-3′ | 380 | |||
| PLM_CytB_SF_R3 | 5′-ATCAGGAATAGGGTGAGTACAC-3′ | |||||
| 4th pair | PLM_CytB_SF_F1 | 5′-TTAATTGACCTCCCCTCAC-3′ | 265 | |||
| PLM_CytB_SF_R4 | 5′-CATTGGCATGGATAGTTCGG-3′ |