- P-ISSN2765-2203
- E-ISSN2765-2211
- KCI Candidate

3권 4호
초록
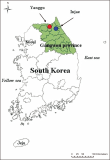
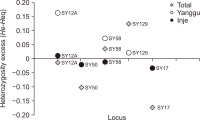
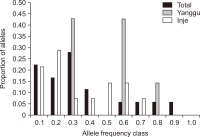
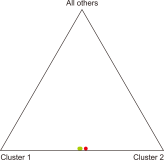
Natural habitats of the Korean long-tailed goral (Naemorhedus caudatus) have been fragmented by anthropogenic activities in South Korea in the last decades. Here, the individual identity, genetic variation, and population differentiation of the endangered species were examined via the multiple-tube approach using a non-invasive genotyping method. The average number of alleles was 3.16 alleles/locus for the total population. The Yanggu population (1.66) showed relatively lower average number of alleles than the Inje population (3.67). Of the total 19 alleles, only seven (36.8%) alleles were shared by the two populations. Using five polymorphic out of six loci, four and six different goral individuals from the captive Yanggu (n=24) and the wild Inje (n=28) population were identified, respectively. The allele distribution was not identical between the two populations (Fisher’s exact test: P<0.01). A considerably low migration rate was detected between the two populations (no. of migrants after correction for size=0.294). Additionally, the F statistics results indicated significant population differentiation between them, however, quite low ( FST=0.327, P<0.01). The posterior probabilities indicated that the two populations originated from a single panmictic population (P=0.959) and the assignment test results designated all individuals to both populations with nearly equal likelihood. These could be resulted from moderate population differentiation between the populations. No significant evidence supported recent population bottleneck in the total Korean goral population. This study could provide us with useful population genetic information for conservation and management of the endangered species.’
Abstract
Natural habitats of the Korean long-tailed goral (Naemorhedus caudatus) have been fragmented by anthropogenic activities in South Korea in the last decades. Here, the individual identity, genetic variation, and population differentiation of the endangered species were examined via the multiple-tube approach using a non-invasive genotyping method. The average number of alleles was 3.16 alleles/locus for the total population. The Yanggu population (1.66) showed relatively lower average number of alleles than the Inje population (3.67). Of the total 19 alleles, only seven (36.8%) alleles were shared by the two populations. Using five polymorphic out of six loci, four and six different goral individuals from the captive Yanggu (n=24) and the wild Inje (n=28) population were identified, respectively. The allele distribution was not identical between the two populations (Fisher’s exact test: P<0.01). A considerably low migration rate was detected between the two populations (no. of migrants after correction for size=0.294). Additionally, the F statistics results indicated significant population differentiation between them, however, quite low ( FST=0.327, P<0.01). The posterior probabilities indicated that the two populations originated from a single panmictic population (P=0.959) and the assignment test results designated all individuals to both populations with nearly equal likelihood. These could be resulted from moderate population differentiation between the populations. No significant evidence supported recent population bottleneck in the total Korean goral population. This study could provide us with useful population genetic information for conservation and management of the endangered species.’
초록
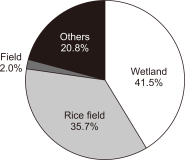
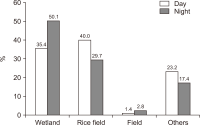
The spring home range and habitat use of the spot-billed duck in Korea were studied using GPS-mobile phone-based telemetry (WT-300). The study areas were Anseong-si, Seosan-si, Nonsan-si, and Sejong-si. Analysis was performed using minimum convex polygon (MCP) and kernel density estimation (KDE) spot-billed ducks had an average home range of 70.28 km² (standard deviation [SD]=84.50, n=6), and a core habitat (50%) 2.66 km² (SD=2.60, n=6), according to MCP and KDE, respectively. Wetlands (41.5%) and rice fields (35.7%) were highly used as habitats. The rice field use rate was high during the day, and the wetland utilization rate was high at night. Rice fields and wetlands were the primary habitats in spring.
Abstract
The spring home range and habitat use of the spot-billed duck in Korea were studied using GPS-mobile phone-based telemetry (WT-300). The study areas were Anseong-si, Seosan-si, Nonsan-si, and Sejong-si. Analysis was performed using minimum convex polygon (MCP) and kernel density estimation (KDE) spot-billed ducks had an average home range of 70.28 km² (standard deviation [SD]=84.50, n=6), and a core habitat (50%) 2.66 km² (SD=2.60, n=6), according to MCP and KDE, respectively. Wetlands (41.5%) and rice fields (35.7%) were highly used as habitats. The rice field use rate was high during the day, and the wetland utilization rate was high at night. Rice fields and wetlands were the primary habitats in spring.

초록
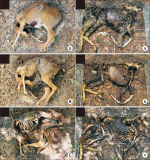
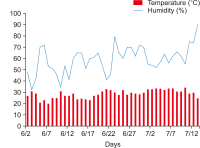
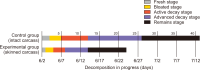
Forensic entomology is a field of study that includes the succession of insects attracted to and found on cadavers. One of its main focusses is estimating post-mortem interval (PMI) based on the growth stage of insects found in and around human cadavers. In many countries, the diversity of insect occurrence is studied in relation to the environmental conditions a cadaver may be exposed to or the effects of different clothes. In this study, changes in the decomposition process and differences in insect succession were investigated by comparing skinned and intact water deer carcasses. Five orders, 15 families, and 21 species of insects were identified, most of which were Dipteran and Coleopteran. The skinned carcass decomposed more rapidly than the intact carcass, which was linked to differences in insect succession. The difference in the decomposition rate and insect succession according to the external conditions of the carcass can be used as basic data for estimating the PMI of the carcass and setting the forensic entomological indicator species.
Abstract
Forensic entomology is a field of study that includes the succession of insects attracted to and found on cadavers. One of its main focusses is estimating post-mortem interval (PMI) based on the growth stage of insects found in and around human cadavers. In many countries, the diversity of insect occurrence is studied in relation to the environmental conditions a cadaver may be exposed to or the effects of different clothes. In this study, changes in the decomposition process and differences in insect succession were investigated by comparing skinned and intact water deer carcasses. Five orders, 15 families, and 21 species of insects were identified, most of which were Dipteran and Coleopteran. The skinned carcass decomposed more rapidly than the intact carcass, which was linked to differences in insect succession. The difference in the decomposition rate and insect succession according to the external conditions of the carcass can be used as basic data for estimating the PMI of the carcass and setting the forensic entomological indicator species.







초록
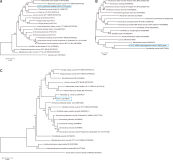
DNA markers have been studied and used intensively to identify plant species based on molecular approaches. The genus Medicago belongs to the family Fabaceae and contains 87 species distributed from the Mediterranean to central Asia. Five species of Medicago are known to be distributed in South Korea; however, their morphological characteristics alone cannot distinguish the species. In this study, we analyzed the phylogenetic relationships using collected five species of Medicago from South Korea and 44 taxa nucleotide information from NCBI. The constructed phylogenetic tree using gibberellin 3-oxidase 1 and tRNALys (UUU) to maturase K gene sequences showed the monophyly of the genus Medicago, with five species each forming a single clade. These results suggest that there are five species of Medicago distributed in South Korea. In addition, we designed polymerase chain reaction primers for species-specific detection of Medicago by comparing the plastid sequences. The accuracy of the designed primer pairs was confirmed for each Medicago species. The findings of this study provide efficient and novel species identification methods for Medicago, which will assist in the identification of wild plants for the management of alien species and living modified organisms.
Abstract
DNA markers have been studied and used intensively to identify plant species based on molecular approaches. The genus Medicago belongs to the family Fabaceae and contains 87 species distributed from the Mediterranean to central Asia. Five species of Medicago are known to be distributed in South Korea; however, their morphological characteristics alone cannot distinguish the species. In this study, we analyzed the phylogenetic relationships using collected five species of Medicago from South Korea and 44 taxa nucleotide information from NCBI. The constructed phylogenetic tree using gibberellin 3-oxidase 1 and tRNALys (UUU) to maturase K gene sequences showed the monophyly of the genus Medicago, with five species each forming a single clade. These results suggest that there are five species of Medicago distributed in South Korea. In addition, we designed polymerase chain reaction primers for species-specific detection of Medicago by comparing the plastid sequences. The accuracy of the designed primer pairs was confirmed for each Medicago species. The findings of this study provide efficient and novel species identification methods for Medicago, which will assist in the identification of wild plants for the management of alien species and living modified organisms.


초록
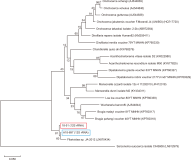
A filarial nematode was found in a blood sample of an Anas falcata individual collected in South Korea in 2018. Phylogenetic analysis based on partial cytochrome C oxidase subunit I (COI) sequences placed the nematode as a novel genus of the family Onchocercidae and as closely related to Mansonella species, Chandlerella quiscali, and filarial nematodes recently reported in avian species. However, different phylogenetic relationship was observed in the NADH dehydrogenase subunit 5 and 12S rRNA-based phylogenetic trees, which might indicate the filarial nematode found in this study was not defined to belong to the known specific genera of the family Onchocercidae. The screening of 105 additional avian blood samples retrieved only one 12S rRNA-targeting polymerase chain reaction (PCR)-positive sample, which indicates that filarial nematode infection is rare in wild birds or that it occurs below the detection limit of PCR in blood samples. Nevertheless, considering the recent findings about ancient interactions between birds and human pathogenic filarial nematodes and their pathogenic potential in several avian species, additional exploration of novel filarial nematodes in wild birds remains necessary.
Abstract
A filarial nematode was found in a blood sample of an Anas falcata individual collected in South Korea in 2018. Phylogenetic analysis based on partial cytochrome C oxidase subunit I (COI) sequences placed the nematode as a novel genus of the family Onchocercidae and as closely related to Mansonella species, Chandlerella quiscali, and filarial nematodes recently reported in avian species. However, different phylogenetic relationship was observed in the NADH dehydrogenase subunit 5 and 12S rRNA-based phylogenetic trees, which might indicate the filarial nematode found in this study was not defined to belong to the known specific genera of the family Onchocercidae. The screening of 105 additional avian blood samples retrieved only one 12S rRNA-targeting polymerase chain reaction (PCR)-positive sample, which indicates that filarial nematode infection is rare in wild birds or that it occurs below the detection limit of PCR in blood samples. Nevertheless, considering the recent findings about ancient interactions between birds and human pathogenic filarial nematodes and their pathogenic potential in several avian species, additional exploration of novel filarial nematodes in wild birds remains necessary.